Project 2. Protein Synthesis

Introduction
The goal of this project is to write a program that mimics the process of protein synthesis in eukaryotic cells. The first half focuses on transcription and translation. The second half introduces the concept of mutation.
Background Information
All living organism store their genetic information in chains of nucleic acid. All eukaryotes (i. e. Organisms whose cells contain membrane-bound organelles, or “little organs”) use deoxyribonucleic acid, or DNA, as the “hard drive” where information is stored. DNA is composed of four distinct nucleobases: adenine, thymine, cytosine, and guanine, which are abbreviated by their first letter (A, T, C, G). A chain of nucleobases form a DNA strand. Although one strand is enough to store information, each eukaryotic cell contains two complementary copies that bind to each other to form a double helix. The rules of base pairing are as follows: A and T pair together, C and G pair together.

Fun Fact: The human genome contains roughly 2.9 billion base pairs. If unwound in a straight line, this would amount to about 2 m in length. Thanks to ingenious folding techniques, our cells are able to store DNA in their nucleus, which is only 6 microns across (1 micron is a millionth of a meter). As if this weren’t impressive enough, remember that each cell contains two strand of DNA!
Task A: Transcription
Each gene codes for a protein, and transcription is the first step of gene expression. Most protein synthesis occurs in organelles known as ribosomes, which are located outside of the nucleus where DNA is stored. To relay information to a ribosome, the cell makes a copy of the relevant gene from DNA and sends that copy out of the nucleus. The copy is called a messenger ribonucleic acid, or mRNA. Like DNA, mRNA is made of the same nucleobases, except for one: it does not contain thymine [T], but instead contains uracil [U]. That means that the complement of [A] in mRNA is [U]. As such, the rules of complementation in mRNA are as follows:
- [A] becomes [U]
- [T] becomes [A]
- [C] becomes [G]
- [G] becomes [C]
Your task is to write a program called transcriptase.cpp that reads a text file called dna.txt that contains one DNA strand per line, which looks as follows:
AAGATGCCG
ATGCCGTAAGATGCGGTAAGATGC
CCGTAAGATGCCGTA
. . .
and outputs to the console (terminal) the corresponding mRNA strands. Each output line must contain exactly one mRNA strand. This is a sample output of the program:
$ ./transcriptase
UUGUACGGC
UACGGCAUUCUACGCCAUUCUACG
GGCAUUCUACGGCAU
. . .
Recall that to read from a file, the following code snipet can be used:
ifstream fin("dna.txt");
if (fin.fail()) {
cerr << "File cannot be read, opened, or does not exist.\n";
exit(1);
}
string strand;
while(getline(fin, strand)) {
cout << strand << endl;
}
fin.close();
The best way to do this is in two steps. First create a function that gives the complement of a base, and then write another function that uses it iteratively over a whole strand.
For example, we could have char DNAbase_to_mRNAbase(char) to return the complement of a base and string DNA_to_mRNA(string) that uses it for each base in the strand. Note that the output must be in capital letters, regardless of how the input is formatted. To do this, you may include the <cstdlib> and use int toupper(int c), which returns the upper case of any alpha character passed to it.
Task B: Translation

While a nucleotide is the basic unit of information, three nucleotides, or codon, is the basic unit of storage. The reason for this is that each gene codes for a protein, and all proteins are made from 20 amino acids. Recall that there are 4 different bases that make up dna. Thus, three bases can encode for 4x4x4 = 64 different symbols. Two base pairs can only encode for 4x4 = 16 symbols, which is not enough.

For this task, you will need the following dictionary file: codons.tsv.
It contains 64 lines, each with two columns. In the first column are the codons, and in the second are the corresponding amino acid.
Your task is to write a program called translatase.cpp that given strands of DNA (taken from dna2b.txt), outputs to the console the corresponding amino-acid chain. Feel free to use your code from Task A to convert the DNA into mRNA to match the codons in the dictionary. Notice that there are 4 special codons: “Met”, which stands for Methionine, and 3 “Stop” codons. Methionine is the first amino acid in every protein chain and as such serves as the “Start” codon. This means that translation does not begin until the “AUG” codon, which encodes for methionine, has been read. The three Stop codons, UAA, UGA, and UAG, are not included in the protein chain and simply signify the end of translation.
The rules of formatting are as follows:
- Use the three-letter abreviation from the dictionary for each amino acid
- Insert a hyphen after each amino acid except for the last
- The first amino acid should always be “Met”
- “Stop” codons should not be inserted
e. g.
tacaacactwould produceMet-Leu.
For this task, you will need to have two ifstream objects open. One for the dna file, and one for the dictionary of codons file. The same code segment from Task A can be adapted to read dna2b.txt since we only read it once. However, for each codon in each of the DNA strand, we need to perform a dictionary lookup. It would not be very efficient to open, read, and close the file each time. The reason is because repetitive file access can become expensive and slow in the long run. The better alternative is to open the file once with one ifstream object, pass it by reference, and reset the file pointer to the beginning for each look up. This can be done with seekg(0). Below is an example that shows how to read from a file that has two fields per line where the delimiter is a space. You can modify this code to perform a look-up in codons.tsv.
void dictionary_read(ifstream &dict) {
string key, value;
dict.clear(); // reset error state
dict.seekg(0); // return file pointer to the beginning
while (dict >> key >> value) {
cout << "key: " << key << endl;
cout << "value: " << value << endl;
}
}
N. B. To make this task a bit easier, the DNA strands are multiples of 3, and must be read as such. This means that you do not need to scan a strand one base at a time until the first AUG. Rather, scan it three bases at a time from the begining of the strand, and start translation at the first AUG encountered in this manner.
Background Information: Mutations
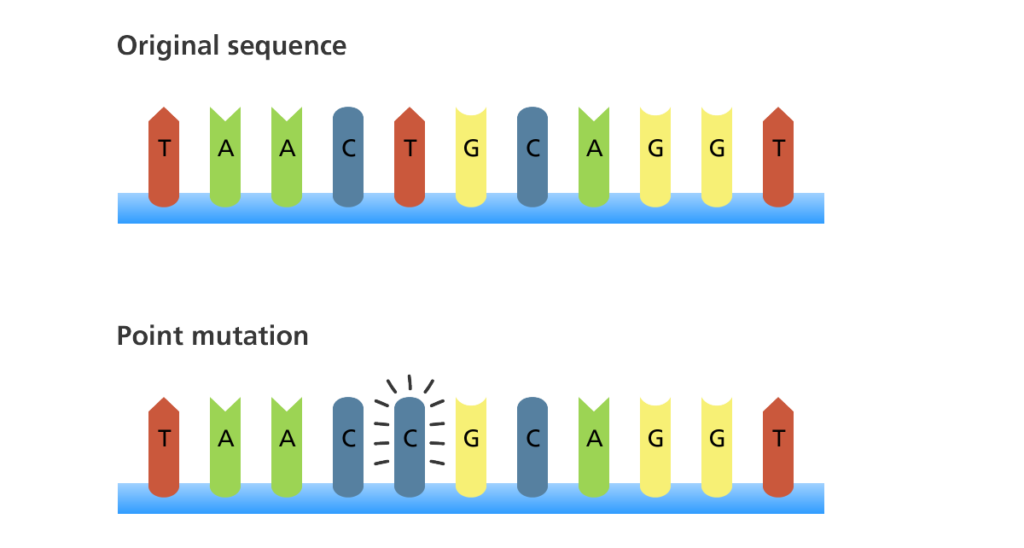
Many factors, such as environmental condition, random chance, and errors in handling, can result in a change, or mutation, in the DNA sequence. These changes can range from benign to catastrophic depending on their effects. There are four kinds of mutations.
- Substitution: a base pair is replaced with another (e. g. aac -> agc).
- Insertion: a new base pair is added somewhere in the DNA strand.
- Deletion: a base pair is removed from the sequence.
- Frameshift: any of the above mutation, or combination thereof, that causes codons to be parsed incorrectly.
Task C: Substitution and Hamming Distance
For this task, we will explore mutations that occur by substitution. Your task is to write a program called hamming.cpp that calculates the Hamming distance between two strings. Given two strings of equal length, the Hamming distance is the number of positions at which the two strings differ.
e. g.: Hamming("aactgc", "atcaga") would output 3.
Notice that certain amino acids are encoded by multiple codons. Therefore, not all substitutions result in a change of protein structure. The file mutations.txt contains an even number of lines (zero-indexed). The even-numbered lines contain the original DNA sequence, and the odd-numbered lines contain that same sequence with substitution mutations. For each pair in mutations.txt, output to the console the Hamming distance followed by “yes” or “no” whether the substitution caused a change in structure.
Example:
$ ./hamming
0 no
17 yes
4 yes
Remember that translation to proteins does not begin until the first “Start” codon, and stops at the first “Stop” codon, and unlike the “Start” codon, the “Stop” codon is not included in the protein chain translation; it simply signifies the end of translation.
Task D: Insertion, Deletion, and Frameshift
The worst type of mutation is the frameshift mutation, as it causes the DNA sequence to be parsed incorrectly. This is often created by a deletion or insertion that causes the sequence to be read in a different multiple of three. This abnormal reading often results in an earlier or later “Stop” codon, which causes the protein to be abnormally short or long, thus rendering it not functional.
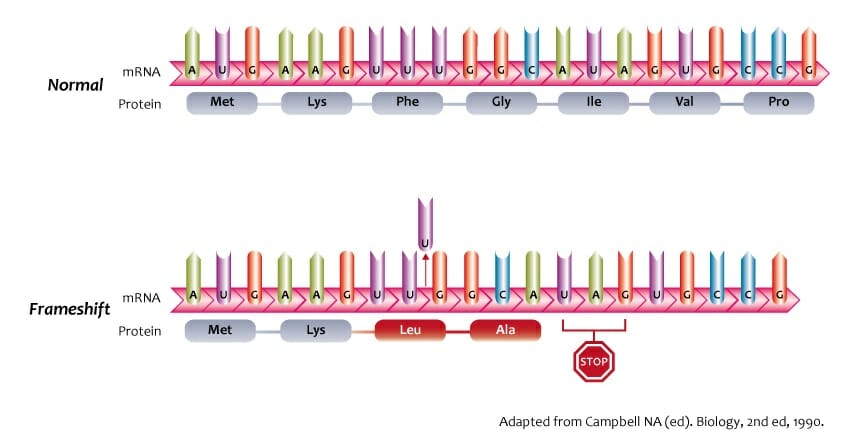
So far, the codons in DNA sequences have been multiples of 3. The file frameshift_mutations.txt contains the same DNA sequences of Task B on the even lines, with frameshift mutations on the odd lines (0-indexed). Each mutation has at most one insertion or one deletion. Your task is to write a program called frameshift.cpp that compares the results of Task B with the mutated strands.
To do this, you will need to parse the strands one nucleotide at a time as the “Start” codon is not a guaranteed multiple of 3 from the begining.
Your output should be the original protein on the even lines, and the mutated protein on the odd lines.
Example:
$ ./frameshit
Met-Thr-Pro-Tyr-Val-Val
Met-Thr-Pro
Met-Gly-Gly-Leu-Tyr
Met-Gly-Gly-Leu-Tyr
Met-Gly-Thr-Ala-Ala-Asp-Pro-Arg-Arg-Gly
Met-Gly-Thr-Ala-Ala-Asp-Ala-Lys-Ala-Gly-Leu